A Doença de Wilson é uma condição genética rara que provoca acúmulo excessivo de cobre no organismo, especialmente no fígado, cérebro e olhos. Esse excesso causa danos progressivos aos órgãos e pode gerar sintomas como alterações hepáticas, neurológicas e psiquiátricas.
Fisiopatologia e causas da Doença do Wilson
Na Doença de Wilson, uma mutação no gene ATP7B compromete a produção da proteína responsável por transportar o cobre para a bile e incorporá-lo à ceruloplasmina. Sem essa função, o cobre não é excretado adequadamente, acumulando-se inicialmente no fígado, onde provoca lesão celular e inflamação.Com o tempo, o excesso de cobre transborda para a circulação, depositando-se no sistema nervoso central, causando sintomas motores, cognitivos e comportamentais e na córnea, formando o anel de Kayser-Fleischer. A doença evolui em fases, começando com alterações hepáticas silenciosas, passando por manifestações neurológicas e psiquiátricas, e, se não tratada, culminando em falência hepática e comprometimento neurológico grave.
Quais são os sintomas da Doença de Wilson?
Manifestações hepáticas
A doença pode causar hepatite crônica, icterícia, cirrose, ascite, varizes esofágicas e hemorragias digestivas. Esses sinais resultam do acúmulo de cobre e do dano progressivo ao fígado, que compromete a função hepática e a circulação portal.
Manifestações neurológicas e psiquiátricas
Incluem tremores, disartria (dificuldade para articular palavras), distonia (movimentos e posturas anormais) e alterações de coordenação. No campo psiquiátrico, podem surgir depressão, psicose, irritabilidade e mudanças de personalidade, que muitas vezes precedem os sintomas motores.
Manifestações oftalmológicas e hematológicas
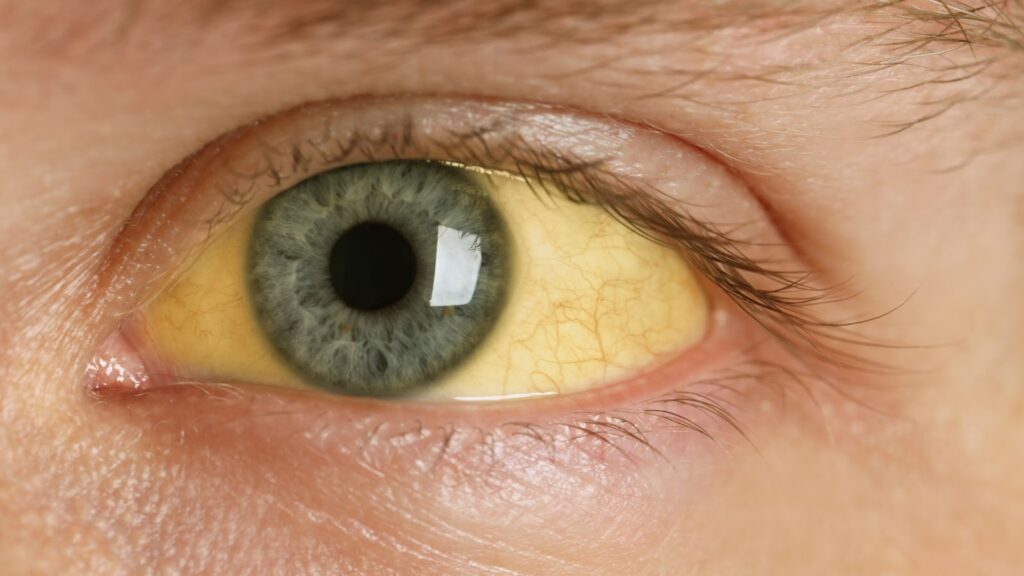
O achado mais típico é o anel de Kayser-Fleischer, um depósito de cobre na córnea visível ao exame com lâmpada de fenda. Outra alteração possível é a catarata em girassol, caracterizada por opacidades radiadas no cristalino. No sangue, pode haver anemia hemolítica, além de alterações laboratoriais como níveis baixos de ceruloplasmina e aumento de cobre urinário.
Como realizar o diagnóstico da Doença de Wilson?

Exames laboratoriais iniciais
A dosagem de ceruloplasmina sérica costuma ser baixa (<20 mg/dL) na maioria dos casos, mas pode ser normal em até 10% dos pacientes. O cobre sérico total geralmente está reduzido, enquanto o cobre urinário em 24 horas está aumentado (>100 µg/24h em adultos), sendo um dos marcadores mais úteis. Esses exames devem ser interpretados em conjunto, pois cada um isoladamente tem limitações.
Exame oftalmológico
O exame com lâmpada de fenda permite identificar o anel de Kayser-Fleischer, presente principalmente em casos neurológicos. Embora não exclusivo da doença, sua presença reforça o diagnóstico quando associada a alterações laboratoriais.
Testes genéticos
A pesquisa de mutações no gene ATP7B confirma o diagnóstico e é especialmente útil para rastrear familiares assintomáticos. No entanto, o exame ainda não está amplamente disponível e pode ser custoso.
Biópsia hepática
É indicada quando o diagnóstico permanece incerto após exames não invasivos. Permite medir diretamente o teor de cobre no fígado (>250 µg/g de tecido seco) e avaliar o grau de inflamação ou fibrose, auxiliando no planejamento terapêutico.
Como é o tratamento da Doença de Wilson?
O tratamento busca remover o excesso de cobre e prevenir seu acúmulo, sendo indispensável por toda a vida para evitar a progressão da doença. As principais abordagens incluem:
- Quelantes de cobre
- Penicilamina: aumenta a excreção urinária de cobre. Pode causar reações alérgicas, síndrome nefrótica, alterações hematológicas e sintomas neurológicos iniciais pela mobilização do cobre.
- Trientina: também promove a excreção de cobre, com menos efeitos adversos que a penicilamina, sendo indicada para pacientes intolerantes a esta. Pode causar anemia por deficiência de ferro.
- Sais de zinco
- Reduzem a absorção intestinal de cobre e são usados em casos assintomáticos, na manutenção após quelantes ou na prevenção em familiares portadores.
- Reduzem a absorção intestinal de cobre e são usados em casos assintomáticos, na manutenção após quelantes ou na prevenção em familiares portadores.
- Dieta com restrição de cobre
- Evitar alimentos ricos em cobre, como fígado, marisco, nozes, chocolate e cogumelos.
- Evitar alimentos ricos em cobre, como fígado, marisco, nozes, chocolate e cogumelos.
- Transplante de fígado
- Indicado em casos de insuficiência hepática fulminante ou doença hepática avançada não controlada com medicação.
O manejo deve ser individualizado e acompanhado por especialista, com monitoramento periódico para ajustar doses e prevenir efeitos colaterais.

Como é a evolução e o prognóstico da Doença de Wilson?
Sem tratamento, a Doença de Wilson progride de forma inevitável, levando a danos irreversíveis no fígado, no sistema nervoso central e em outros órgãos. Isso pode resultar em insuficiência hepática, incapacidade neurológica grave e morte precoce.
Com diagnóstico precoce e adesão rigorosa ao tratamento, a expectativa de vida se aproxima da população geral, e muitos pacientes conseguem manter boa qualidade de vida. O controle adequado impede o acúmulo de cobre e pode até reverter alguns sintomas iniciais.
Nos casos em que há necessidade de transplante hepático, o prognóstico costuma ser favorável, com alta taxa de sobrevida e melhora significativa das manifestações hepáticas. No entanto, alterações neurológicas avançadas podem persistir.
Por fim, a adesão terapêutica é fundamental, pois a interrupção do tratamento favorece a reativação da doença, podendo causar descompensação rápida e potencialmente fatal.
Quais são os principais diagnósticos diferenciais?
A Doença de Wilson pode ser confundida com diversas condições que apresentam sintomas hepáticos, neurológicos ou psiquiátricos semelhantes. Entre os principais diagnósticos diferenciais estão:
- Hepatite autoimune: Causa inflamação hepática crônica, mas geralmente apresenta autoanticorpos específicos (ANA, anti–SMA) e níveis normais de cobre urinário.
- Hemocromatose: Doença de sobrecarga de ferro, com ferro sérico elevado e saturação de transferrina aumentada, diferindo do acúmulo de cobre.
- Esclerose múltipla: Pode simular sintomas neurológicos, porém há lesões características na ressonância magnética e ausência de sintomas hepáticos.
- Distonias: Apresentam alterações motoras semelhantes às neurológicas da Doença de Wilson, mas sem comprometimento hepático ou alterações laboratoriais típicas.
- Psicose juvenil: Pode apresentar sintomas psiquiátricos semelhantes, porém sem sinais físicos ou laboratoriais que indiquem acúmulo de cobre.
Desse modo, a avaliação detalhada clínica, laboratorial e de imagem é essencial para diferenciar essas doenças e conduzir ao diagnóstico correto.
Quando suspeitar de Doença de Wilson?
A suspeita deve surgir principalmente em pacientes jovens (entre 5 e 35 anos) que apresentam sinais de comprometimento hepático inexplicado, sintomas neurológicos progressivos ou alterações psiquiátricas sem causa definida.
Considera-se a doença em casos de:
- Hepatite crônica, cirrose ou insuficiência hepática em idade precoce;
- Tremores, distonia, disartria ou outras manifestações neurológicas sem diagnóstico claro;
- Alterações psiquiátricas recentes, como depressão, irritabilidade ou psicose, especialmente associadas a sintomas motores;
- Presença do anel de Kayser-Fleischer ao exame oftalmológico;
- Anemia hemolítica de causa desconhecida ou alterações laboratoriais sugestivas de acúmulo de cobre.
A investigação precoce permite diagnóstico e tratamento eficaz, melhorando o prognóstico do paciente.
Como evitar a doença de Wilson?
A Doença de Wilson é uma condição genética, portanto não há prevenção possível. O diagnóstico precoce e o tratamento adequado são essenciais para evitar suas complicações.
Como revisar Doença de Wilson para provas de residência com a MedCof?
A MedCof oferece uma preparação completa, que vai além das questões de prova. Com foco nos pilares teóricos, clínicos e práticos, você aprende os conceitos essenciais da Doença de Wilson e como aplicá-los na rotina médica.
Essa abordagem sólida facilita o entendimento, ajuda na fixação do conteúdo e prepara para os desafios da residência e da prática profissional. Assim, você chega mais confiante e preparado para todas as etapas do seu caminho.
