
O domínio das epilepsias na faixa etária pediátrica exige do médico residente a capacidade de integrar a neurofisiologia, a genética e a clínica. As provas de residência médica focam não apenas no manejo da crise aguda, mas no reconhecimento de síndromes eletroclínicas específicas e suas bases etiológicas!
Definições e Classificação das Epilepsias (ILAE 2017)
A distinção precisa entre os termos é o primeiro passo para o diagnóstico correto e é frequentemente explorada em questões teóricas.
- Crise Epiléptica: Define-se como a ocorrência transitória de sinais e/ou sintomas decorrentes de atividade neuronal excessiva ou síncrona no cérebro.
- Epilepsia: Doença caracterizada pela predisposição persistente do cérebro em gerar crises. O diagnóstico clínico é estabelecido por um dos seguintes critérios:
- Duas crises não provocadas (ou reflexas) com intervalo superior a 24 horas.
- Uma crise não provocada associada a uma probabilidade de recorrência igual ou superior a 60% (ex: presença de alteração estrutural em neuroimagem ou atividade epileptiforme no EEG).
- Diagnóstico de uma síndrome epiléptica específica.
Conceito de Epilepsia Resolvida: Aplica-se a indivíduos que ultrapassaram a idade de uma síndrome idade-dependente ou que permanecem sem crises há 10 anos, estando fora de uso de fármacos anticrise há pelo menos 5 anos.
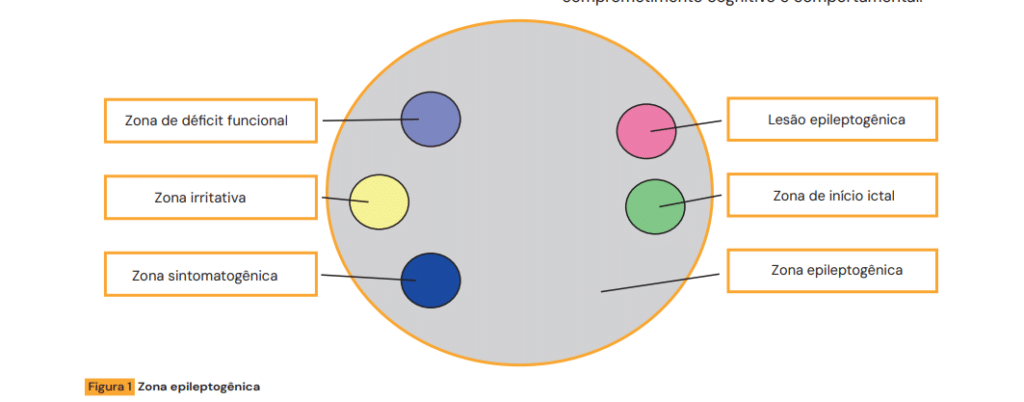
Semiologia Ictal e Topografia Lesional
A análise detalhada da semiologia permite inferir a Zona Epileptogênica (região responsável pela geração das crises). Em provas, a descrição clínica da crise é a chave para identificar o lobo acometido.
Epilepsias do Lobo Temporal
Caracterizam-se geralmente por início progressivo e pós-ictal prolongado.
- Temporal Mesial: Envolve estruturas do sistema límbico (amígdala, hipocampo). Manifesta-se com auras epigástricas ascendentes, sintomas emocionais (medo, déjà vu, jamais vu) e automatismos orofaciais/manuais. Um sinal lateralizatório clássico cobrado em provas é a postura distônica do membro superior contralateral à zona epileptogênica, enquanto o paciente apresenta automatismos ipsilaterais (como roçar o nariz).
- Temporal Lateral: Predominam alucinações auditivas, ilusões visuais ou vertigem. A evolução para crise tônico-clônica bilateral é mais frequente do que na mesial.
Epilepsias do Lobo Frontal
As crises são tipicamente breves, de início e término súbitos, com predomínio durante o sono e rápida recuperação da consciência (pós-ictal curto).
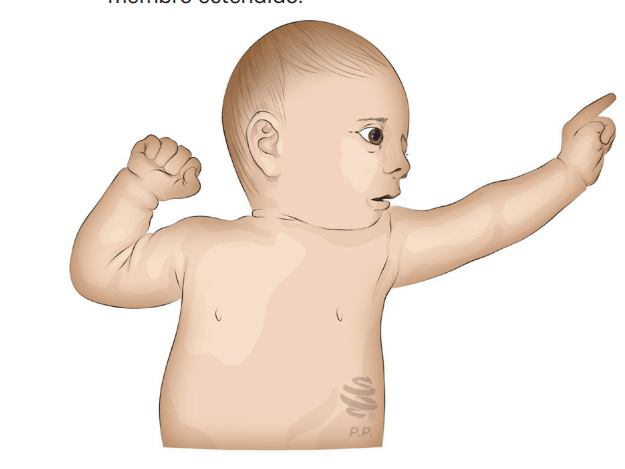
- Área Motora Suplementar: Apresenta a “postura do espadachim” (extensão tônica assimétrica), onde a zona epileptogênica é contralateral ao membro estendido e ipsilateral ao membro fletido.
- Giro Pré-Central: Clonias contralaterais com a clássica marcha Jacksoniana (progressão das clonias seguindo o homúnculo motor).
- Cíngulo Anterior: Automatismos hipermotores, pedalagem, expressão facial de medo e a fácies de chapeau de gendarme (boca invertida).
Epilepsias do Lobo Occipital
Manifestações visuais (alucinações simples como pontos luminosos ou complexas), amaurose ictal, desvio ocular tônico e sintomas autonômicos importantes, como vômitos ictais e cefaleia peri-ictal intensa (mimetizando enxaqueca).
Urgências Neurológicas: Crise Febril e Estado de Mal
Crise Febril
Ocorre entre 6 e 60 meses, na vigência de febre sem infecção do SNC. A distinção é vital:
- Simples: Crise generalizada (tônico-clônica), com duração inferior a 15 minutos e não recorrente em 24h. O prognóstico é benigno e não exige exames complementares (EEG/Neuroimagem) na maioria dos casos.
- Complexa: Focal, duração superior a 15 minutos ou recorrente em 24h. Associa-se a um risco aumentado de desenvolvimento de epilepsia futura.
- Nota: A crise febril não requer tratamento anticonvulsivante contínuo profilático (fenobarbital ou valproato) na vasta maioria dos casos.
Estado de Mal Epiléptico (EME)
Definido operacionalmente como crise com duração igual ou superior a 5 minutos ou crises subentrantes sem recuperação da consciência entre elas. A abordagem terapêutica deve ser escalonada e rápida (“tempo é cérebro”):
- Estabilização (0-5 min): ABC, monitorização, acesso venoso e correção imediata de hipoglicemia (se presente).
- Terapia Inicial (1ª Linha): Benzodiazepínico.
- Diazepam 0,3 mg/kg EV ou Midazolam 0,2 mg/kg EV/IM.
- Na falta de acesso: Diazepam via retal ou Midazolam via nasal/bucal.
- Segunda Linha (persistência da crise): Fenitoína 15-20 mg/kg EV (atenção à velocidade máxima de infusão para evitar arritmias e hipotensão) ou Fosfenitoína.
- Terceira Linha: Fenobarbital 20 mg/kg EV ou Valproato de Sódio EV.
- EME Refratário: Indução de coma anestésico com Midazolam contínuo, Tiopental ou Propofol, necessitando de intubação orotraqueal e monitorização EEG contínua em UTI.
Síndromes Epilépticas Específicas por Faixa Etária
O reconhecimento sindrômico orienta o tratamento (evitando fármacos que pioram o quadro) e define o prognóstico.
Período Neonatal e Lactente (0-2 anos)
- Epilepsias autolimitadas: Frequentemente associadas a canalopatias genéticas (ex: KCNQ2 na Neonatal Familiar, PRRT2 na Infantil). Caracterizam-se por desenvolvimento neuropsicomotor normal e remissão espontânea das crises.
- Síndrome de West (Espasmos Infantis):
- Tríade clássica: Espasmos epilépticos (em flexão ou extensão, em salvas) + Hipsarritmia (padrão caótico de alta voltagem no EEG interictal) + Regressão ou atraso do desenvolvimento.
- Tratamento: Vigabatrina (1ª escolha, especialmente se associado à Esclerose Tuberosa) e Corticosteroides em altas doses (Prednisolona ou ACTH).
- Síndrome de Dravet:
- Encefalopatia epiléptica e do desenvolvimento grave, geralmente associada a mutação no gene SCN1A (canal de sódio). Inicia-se no primeiro ano de vida com crises febris complexas/prolongadas, evoluindo para crises refratárias e declínio cognitivo.
- Alerta de Prova: São contraindicados os bloqueadores de canais de sódio (Carbamazepina, Fenitoína, Lamotrigina), pois podem exacerbar as crises. O tratamento envolve Valproato, Clobazam, Estiripentol ou Canabidiol.
- Síndrome de Ohtahara: Encefalopatia precoce (< 3 meses), caracterizada pelo padrão surto-supressão no EEG e prognóstico neurológico reservado.
Infância
- Epilepsia Rolândica (Autolimitada com Espículas Centrotemporais):
- A epilepsia focal mais comum da infância. Apresenta crises focais motoras ou sensitivas na face/orofaringe, sialorreia, sons guturais e parada da fala, ocorrendo predominantemente durante o sono ou ao despertar. O EEG mostra espículas centrotemporais ativadas pelo sono.
- Prognóstico excelente, frequentemente remitindo na adolescência e muitas vezes dispensando tratamento medicamentoso.
- Síndrome de Panayiotopoulos:
- Epilepsia autolimitada com crises autonômicas. O quadro clínico destaca-se por vômitos ictais recorrentes, palidez e síncope (“ictal syncope”), com crises frequentemente prolongadas (>30 min), mas de prognóstico benigno.
- Síndrome de Lennox-Gastaut:
- Encefalopatia epiléptica grave em pré-escolares.
- Tríade: Múltiplos tipos de crise (tônica noturna, atônica/drop attacks, ausência atípica) + Deficiência Intelectual + EEG com ponta-onda lenta (< 2,5 Hz) difusa.
- Tratamento é desafiador, utilizando combinações como Valproato, Lamotrigina, Clobazam, Rufinamida ou Canabidiol. Drop attacks refratários podem ter indicação de calosotomia.
Adolescência
- Epilepsia Mioclônica Juvenil (EMJ):
- Início típico entre 12-18 anos. Caracteriza-se pela tríade de crises: mioclonias (choques nos membros superiores, derrubando objetos), crises tônico-clônicas generalizadas e ausências. As mioclonias ocorrem tipicamente ao despertar.
- Precipitantes clássicos: Privação de sono, estresse e consumo de álcool.
Tratamento: Ácido Valpróico é a droga de escolha. Em mulheres em idade fértil, deve-se ter cautela devido à teratogenicidade, optando-se por Levetiracetam ou Lamotrigina. Deve-se evitar Carbamazepina e Fenitoína, que podem piorar as mioclonias e ausências.
Outras Encefalopatias e Etiologias Específicas
- Síndrome de Landau-Kleffner: Caracterizada pela agnosia auditiva verbal adquirida (a criança ouve, mas não compreende, simulando surdez) e regressão da linguagem. O EEG revela descargas contínuas durante o sono de ondas lentas (POCS). Requer tratamento agressivo (corticosteroides, benzodiazepínicos) para tentar preservar a função cognitiva.
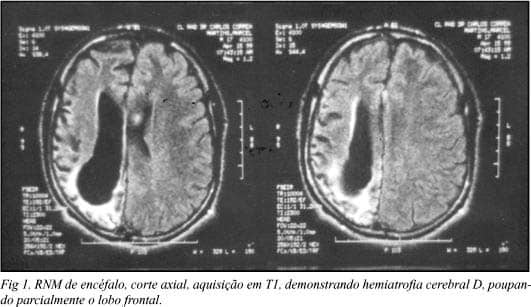
- Encefalite de Rasmussen: Epilepsia focal crônica progressiva de etiologia imunológica. Cursa com crises focais intratáveis (epilepsia partialis continua), hemiparesia progressiva e atrofia de um hemisfério cerebral na neuroimagem. O tratamento definitivo muitas vezes é a hemisferotomia funcional.
- Deficiência do Transportador de Glicose tipo 1 (GLUT-1): Encefalopatia metabólica que cursa com epilepsia de início precoce, microcefalia e distúrbios do movimento que pioram com jejum. O tratamento padrão-ouro é a Dieta Cetogênica, que fornece corpos cetônicos como fonte alternativa de energia cerebral.
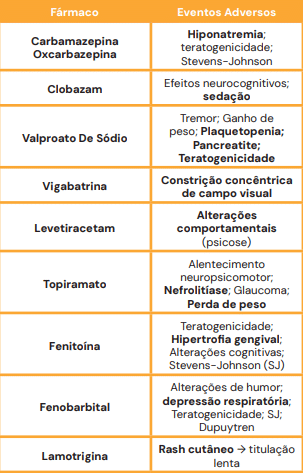
Terapêutica Farmacológica: Mecanismos e Efeitos Adversos
O conhecimento dos efeitos colaterais é frequente alvo de questões.
Conquiste a aprovação e acompanhe mais conteúdos!
Gostou da novidade? Continue acompanhando nosso blog para ficar por dentro das principais notícias sobre residência médica, editais e oportunidades que podem transformar sua carreira!
